The Help Conquer Cancer research team at the Ontario Cancer Institute continues to analyze the millions of protein-crystallization images processed by World Community Grid volunteers, by building new classifiers based on a combination of Grid-processed image features, and deep features learned directly from image pixels. Improvements in image classification, along with new data provided by our collaborators increase possibilities for discovering useful and interesting patterns in protein crystallization.
Since our last Help Conquer Cancer (HCC) project update, we have continued to analyze the results that you generated. Here, we provide an update on that analysis work, and new research directions the project is taking.
Analyzing HCC Results
Volunteers for the HCC project received raw protein crystallization images and processed each image into a set of over 12,000 numeric image features. These features were implemented by a combination of image-processing algorithms, and refined over several generations of image-processing research leading up to the launch of HCC. The features (HCC-processed images) were then used to train a classifier that would convert each image's features into a label describing the crystallization reaction captured in the image.
Importantly, these thousands of features were human-designed. Most protein crystals have straight edges, for example, and so certain features were incorporated into HCC that search for straight lines. This traditional method of building an image classifier involves two types of learning: the crystallographer or image-processing expert (human), who studies the image and designs features, and the classifier (computer model), that learns to predict image labels from the designed features. The image classifier itself never sees the pixels; any improvements to the feature design must come from the human expert.
More recently, we have applied a powerful computer-vision/machine-learning technology that improves this process by closing the feedback loop between pixels, features and the classifier: deep convolutional neural networks (CNNs). These models learn their own features directly from the image pixels; thus, they could complement human-designed features.
CrystalNet
We call our deep convolutional neural networks CrystalNet. Our preliminary results suggest that it is an accurate and efficient classifier for protein crystallization images.
In a CNN, multiple filters act like pattern detectors that are applied across the input image. A single map of the layer 1 feature maps shows the activation responses from a single filter. Deep CNNs refers to CNNs with many layers: higher-level filters stacked upon lower-level filters. Information from image pixels at the bottom of the network rises upwards through layers of filters until the "deep" features emerge from the top. Although the example shown in Figure 1 (below) has only 6 layers, more layers can be easily added. Including other image preprocessing and normalization layers, CrystalNet has 13 layers in total.
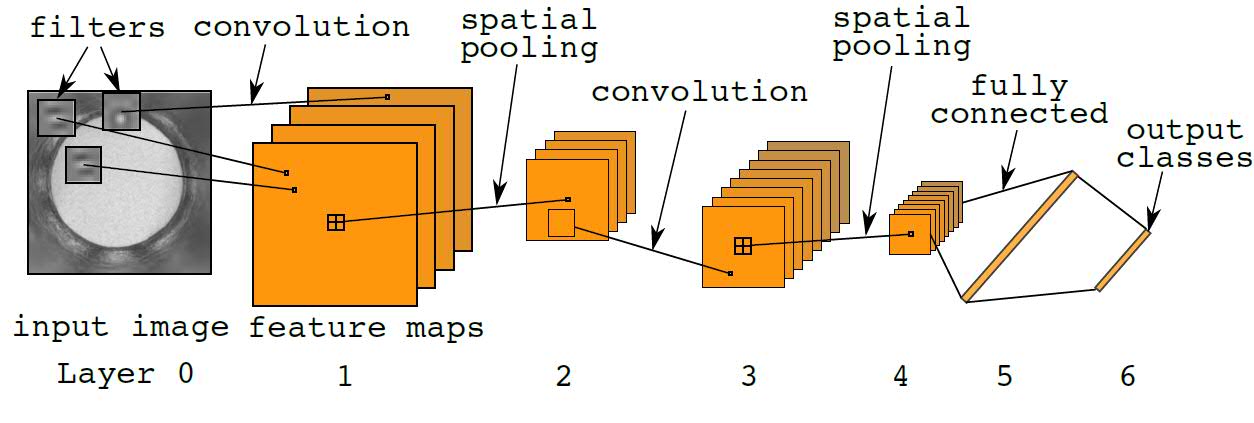
Fig. 1: Diagram of the standard convolutional neural network. For a single feature map, the convolution operation applies inner product of the same filter across the input image. 2D topography is preserved in the feature map representation. Spatial pooling performs image down-sampling of the feature maps by a factor of 2. Fully connected layers are the same as standard neural network layers. Outputs are discrete random variables or "1-of-K" codes. Element-wise nonlinearity is applied at every layer of the network.
After training, Figure 2 (below) shows examples of the first layer filters. These filters extract interesting features useful for protein crystallography classification. Note that some of these filters look like segments of straight lines. Others resemble microcrystal-detecting filters previously designed for HCC.

Fig. 2: Selected examples of the first-layer filters learned by our deep convolutional neural net. These filters have resemblances to human-designed feature extractors such as edge (top row), microcrystal (bottom), texture, and other detectors from HCC and computer vision generally.
Figure 3 (below) shows CrystalNet's crystal-detection performance across 10 image classes in the test set. CrystalNet produces an area under curve (AUC) 0.9894 for crystal class classification. At 5% false positive rate, our model can accurately detect 98% of the positive cases.
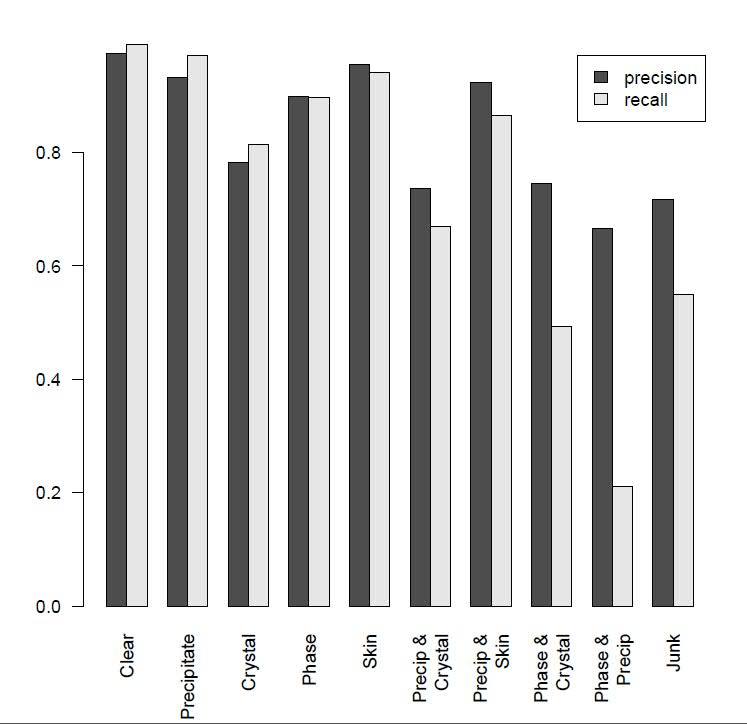
Fig. 3: CrystalNet 10-way image classification performance.
CrystalNet can provide labels for images generated during the high-throughput process effectively, with a low miss rate and high precision for crystal detection. Moreover, CrystalNet operates in real-time, where labeling 1,536 images from a single plate only requires approximately 2 seconds. The combination of accuracy and efficiency makes a fully automated high-throughput crystallography pipeline possible, substantially reducing labor-intensive screening.
New data from collaborators
Our collaborators at the High-Throughput Screening Lab at the Hauptman-Woodward Medical Research Institute (HWI) supplied the original protein-crystallization image data. They continue to generate more, and are using versions of the image classifiers derived from the HCC project.
Our research on the predictive science of protein crystallization has been limited by the information we have about the proteins being crystallized. Our research partners at HWI run crystallization trials on proteins supplied by labs all over the world. Often, protein samples are missing the identifying information that allows us to link these samples to global protein databases (e.g., Uniprot). Missing protein identifiers prevent us from integrating these samples into our data-mining system, and thereby linking the protein's physical and chemical properties to each cocktail and corresponding crystallization response.
Recently, however, HWI crystallographers were able to compile and share with us a complete record of all crystallization-trial proteins produced by the North-Eastern Structural Genomics (NESG) group. This dataset represents approximately 25% of all proteins processed by HCC volunteers on World Community Grid. Now all our NESG protein records are complete with each protein's Uniprot ID, amino-acid sequence, and domain signatures.
With more complete protein/cocktail information, combined with more accurate image labels from improved deep neural-net image classifiers, we anticipate greater success mining our protein-crystallization database. Work is ongoing.