In this update, the Forli Lab team at Scripps Research gives us a look at the three viral proteins they’re currently studying and describes their plans for the near future.
Background
Our team at Scripps Research is doing molecular modeling simulations to look for possible candidates for the development of treatments for COVID-19. To be successful, we’ve partnered with World Community Grid for the massive computing power we need to carry out millions of simulated laboratory experiments.
In this update, we’ll give you all a look at the three viral proteins we’re currently studying, outline our plans to study more proteins with the help of World Community Grid, and let you know where we are in the process of getting potential treatments to our laboratory collaborators for testing. (We’re close!)

But first, research team member Dr. Martina Maritan has created an infographic about the project to help outline the full research process. You can click here to see the full details.
Additionally, here are definitions for a few terms to help you better understand our update.
- Ligand pose: A ligand pose is one of the possible arrangements for a given molecule. As part of this project, we have multiple ligand poses for each chemical compound, and we pick the best one.
- Molecular docking: This process is the study of how two or more molecules fit together, such as how a protein in a human cell or in a virus fits with a chemical compound.
In OpenPandemics - COVID-19, we can leverage this process at scale, thanks to World Community Grid's massive computational power, by virtually screening millions of chemical compounds to see which might be capable of binding to proteins of the SARS-Cov2 virus which causes COVID-19.
- Viral proteins: These are proteins generated by a virus (in this project, by SARS-Cov2).
Data management and analysis
In the previous update we detailed how we managed to compress the results further than possible with traditional data compression by only transmitting each ligand pose’s variables (which we technically refer to as "genome"). This is a short update on what happens after we “rehydrate” those poses, or in other words, how we analyze the results.
The data you all create is analyzed in multiple stages. The first stage happens when we receive packages (or batches of work units) from World Community Grid. We analyze each ligand pose contained in them (each package contains about half a million poses) and capture specific interactions with the protein target. These interactions can be hydrogen bonds, non-polar interactions, or, most importantly for the reactive docking, the occurrence of the chemical reaction between the warheads and the targeted residues.
This information, along with the result genome is what we store in our results database for the next stage of analysis. Currently, through your dedicated effort you have created a total of about 6.8 billion ligand poses taking up about 20 TB of storage. While this is a lot of data - and we did recently update our database server with more storage (250 TB!) for this very reason - it is still only about as much as just the individual coordinates for each pose would require. While this stage of the analysis is the most computationally expensive, we are currently able to analyze about four times as many packages per day than we are receiving using only a single compute node.
The second stage of analysis comprises multiple levels of filtering over the entire set of results, using the interactions we captured in the first phase. We start from a broad filter based on ligand energies and interactions with residues of interest for a given target. This database query returns individual pose identifiers. Those poses are then categorized by which selection criteria they fulfill. These selection criteria are target specific and include things such as “has interactions with x but none with y”, “hydrogen bonds with z”, and others. Finally, ligand poses that meet all of the most important criteria are rehydrated (i.e., their actual 3D coordinates are restored) and stored as potential candidates. Currently, this stage of the analysis yields about 20,000 potential candidates out of the 6.8 billion ligand poses.
The final stage of analysis is to manually select the most promising molecules for experimental testing by our collaborators. The selection process consists of visual inspection of the docked poses to evaluate a few aspects that are necessary for these molecules to bind the protein, but that often are not properly modeled by docking. Shape complementarity is one of these aspects: the molecule should fit nicely in the protein pocket, without excessive empty space between the molecule and the protein. Another important aspect is strain - the molecule shouldn’t make unfavorable interaction with itself in order to fit in the pocket.
Out of the 20,000 potential candidates we selected about 70 molecules for experimental testing and we are going to order them soon! (See the section below on “Making virtual molecules REAL.”)
Current packages
Above: a rendering of the nsp3 protein
>Above: a rendering of the nsp5 protein
Above: a rendering of the nsp15 protein
The current packages are targeting three viral proteins (see the videos above which were created by research team member Dr. Jérome Eberhardt): nsp3, nsp5 and nsp15. This is the first time that molecules are being docked against nsp5 and nsp15.
All molecules contain the acrylamide group that may be capable of reacting with certain cysteines in the viral proteins. All these acrylamide-containing molecules are available in the ZINC database or directly from our chemical suppliers, ensuring that they are reasonably priced or otherwise easy to synthesize.
New targets
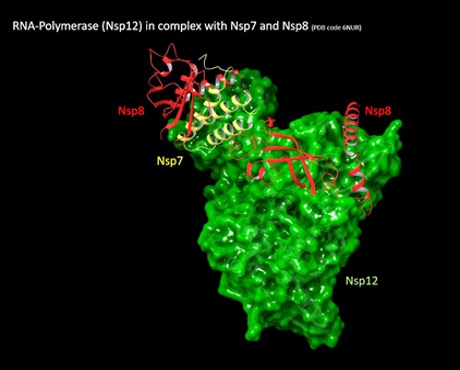
While we keep preparing packages for the current targets, we are already exploring new ones for which we will run the next screenings. One of them is the non-structural protein 8 (nsp8).
Similarly to other coronaviruses, SARS-CoV-2 presents 16 non-structural proteins that are highly conserved and presenting different functions, including the formation of the replication-transcription complex. Among them, Nsp8 is a coronaviral non-structural protein that together with nsp7 interact with and regulate the RNA-dependent RNA-polymerase (nsp12, or RdRp), which catalyses the synthesis of the viral RNA during the infection. Essentially, nsp8 and nsp7 act as co-factors stimulating the polymerase activity of nsp12, playing a key role in the replication and transcription cycle of the virus.
From a structural point of view, nsp8 assumes different conformations depending on the interacting partner, in particular, it is found in a ‘close conformation’ when bound to nsp7 and in a ‘open conformation’ when bound to nsp12. We started our investigation from the nsp7-nsp8-nsp12 ternary complex (Pdb id: 6NUR) focusing at first only on the nsp8 protein at the interface with nsp12, which presents two cysteine residues 114 and 142 respectively.
In collaboration with the Gervasio Lab, we decided to investigate the accessibility of these pockets containing the cysteine residues using aqueous and co-solvent dynamic simulations. Subsequently, a careful selection of nsp8 structures derived from the dynamics will be then used as input targets for virtually screening libraries of reactive fragments.
Making virtual molecules REAL
We are working with the chemical vendor Enamine who will be providing the physical analogs of the molecules that have been docked using the World Community Grid. These molecules come from their REAL database, a set of molecules for which they have a synthetic route planned, but which have not necessarily been synthesized in the past. Their REAL catalog contains 1.36 billion molecules that we can immediately screen and can be synthesized on-demand. This allows us to explore more of the chemical space than we could with existing physical libraries.
This means that soon we’ll be sending off molecules to our collaborators to validate our screening results, and hopefully bring us one step closer to the first inhibitors!
Thanks to everyone who is supporting OpenPandemics – COVID-19!