Mapping cancer markers researchers answer questions asked on the forum
You asked: “Are there any plans to optimize this project for a particular operating system Linux/Windows/ Mac OS/Android ? I think the GPU option was answered a while back as being non-applicable.” and “Are there plans to create a GPU application for MCM, as GPU's are far more powerful than CPU's (see the OPN-project for evidence of this)?”
We do agree that GPUs are more powerful than CPUs (especially for certain computational workloads), and it is indeed applicable to MCM, but at this stage and with running WCG, we do not have the manpower to port our code to GPUs, unfortunately. It is, though, on our (long) todo list.
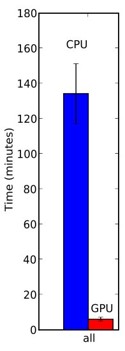
Indeed, our Help Conquer Cancer project was running both CPU and GPU versions. Back in 2009, WCG comprised over 1.4 million devices, and we were getting about 156 CPU/years a day of computation. GPUs required about 6 minutes per image, while CPUs required about 2 hours (testing across a range of CPUs and GPUs).
As for optimizing any WCG code to different architectures and machines - that is a much longer goal and task. We are actively looking for partners to work on optimizing heterogeneous grid computation. With expanded partners and WCG team - this is one of our longer term goals. Related to it is the intention to expand applications that are useful to multiple projects and optimize and BOINC-enable them in advance - so on-boarding new projects that use them is faster.
You asked: “From "One of WCG's programs – Mapping Cancer Markers – identified and validated a diagnostic group of biomarkers for lung cancer, after processing over 9.8 trillion possible biomarkers. Using the various data points identified from the tissue samples and the processing power of WCG, these six markers are now being validated clinically." Could that be expanded upon in a future update?”
Probesets are sets of molecules that bind to specific genes and are used to measure each gene’s expression (biological activity). The MCM project sampled and evaluated 9.8 trillion signatures (combinations of probesets), and evaluated these signatures in a lung cancer diagnostic gene expression dataset collected from lung brushes.
At first, signatures of 5–100 probesets were drawn randomly from the 22,283 available in the dataset. We used preliminary results from the first stage to estimate the frequency of each probeset in top-performing signatures. In the second stage, signatures of 10–25 probesets were drawn randomly from the most frequent 1%..
Using Matthews correlation coefficient (MCC, a statistical measure of quality of a prediction) higher than 0.8 as a performance threshold, over 49 million high-performing signatures have been identified. Of those, six signatures shared the top MCC score. These six signatures comprise 23 to 25 probesets. We are confirming their clinical value by testing their performance on additional 22 independent lung cancer expression datasets. 16 of these datasets measured gene expression in lung biopsies, one in saliva, one in whole blood, one in endobronchial epithelial lining fluid, one in broncheoalveolar lavages, one in peripheral airways brushes and one in bronchial epithelial brushes (like our original dataset). We expect our validation to perform better in the latter 4 datasets because the expression is measured on tissues more similar to (or the same as) the one used in our original dataset.
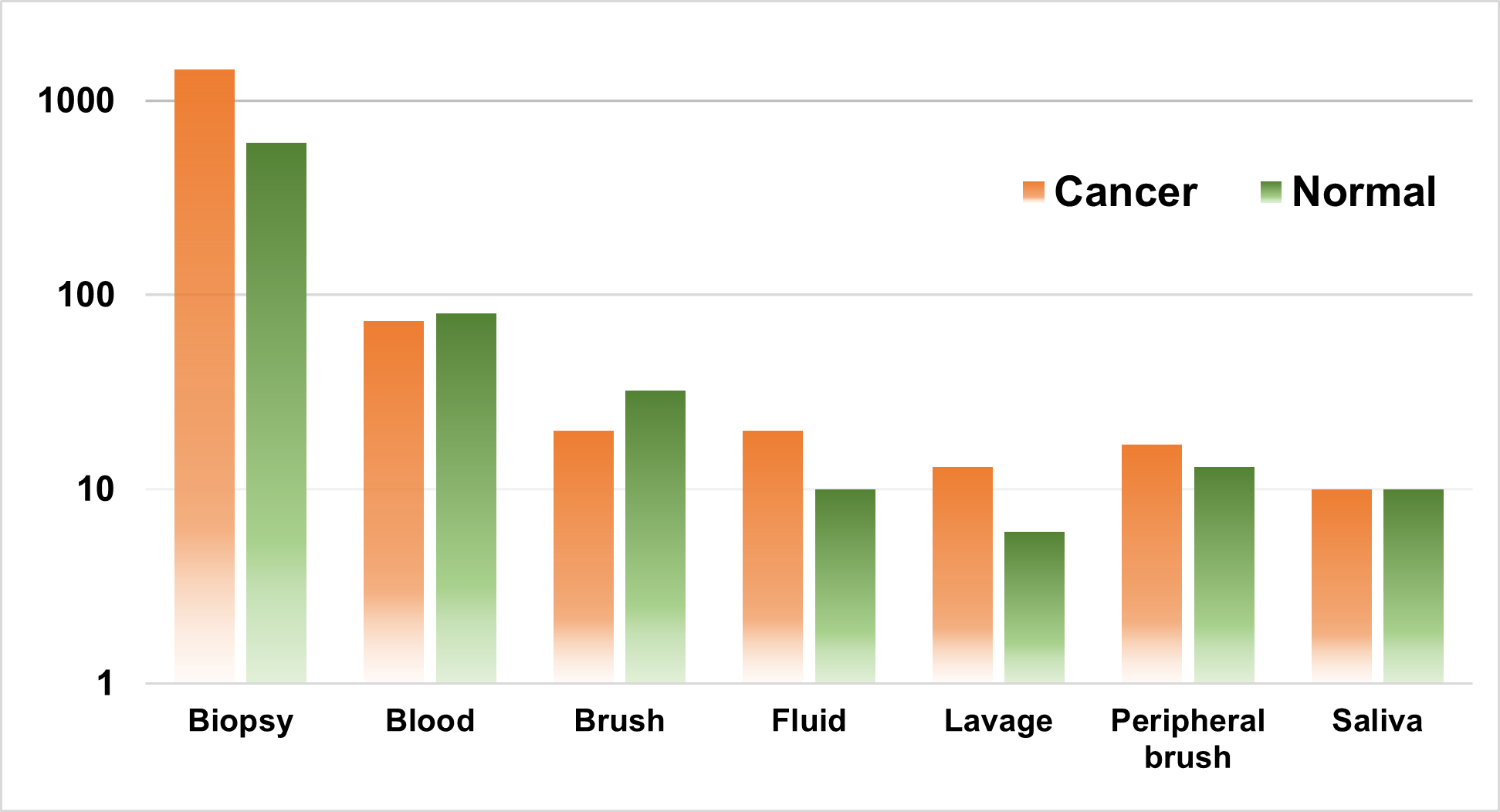
Number of samples (in logarithmic scale) per tissue type. The number of samples from the 16 datasets that measured gene expression in lung biopsies were combined.