Thanks to your help, the Mapping Cancer Markers team is nearly finished with benchmarking their first set of genetic markers. In this update, the team presents an in-depth review of what they've accomplished thus far, and what significance this early work will have for cancer research at their lab and elsewhere.
We are happy to report that we have begun to analyze the results using a high-throughput analytics package to assess the fitness and landscape of gene signature sizes between 5 and 25 genes. This analysis has shown that smaller signatures usually comprise different genes compared to larger signatures (i.e., you cannot "build" a larger signature from small ones), and that those genes are targeting many different signaling cascades and biological processes.
Analytics
To get a better understanding of how much data our team is receiving, we'd like to briefly introduce one of the tools that we have adopted to analyze the incoming results. From the very beginning of the project, it was clear that analyzing such a large, ongoing flow of data would be a challenge. Thus, we started to use the IBM® InfoSphere® Streams real-time analytics platform to streamline the analysis pipeline. When complete, our Streams application will run continuously, processing members' work units in real time as we receive them. We currently have the core analysis framework implemented and running on a subset of the MCM results. We will continue to add additional layers of analysis, and fine-tune our system until it is running at full capacity. For that reason, we have dedicated one of our main compute servers (IBM Power® 780) to analyzing MCM results.
Results
Pictured below is a sampling (a very small fraction) of some of the ongoing work that will establish a benchmark for further experiments. Each dot in both of the graphs is a potential lung-cancer biomarker. These graphics are distilled from thousands of MCM results sent back by World Community Grid members.
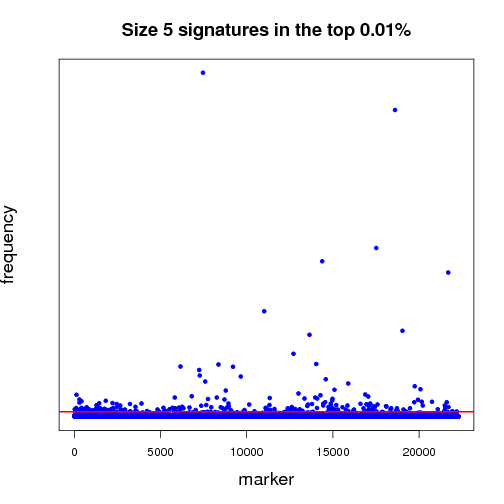
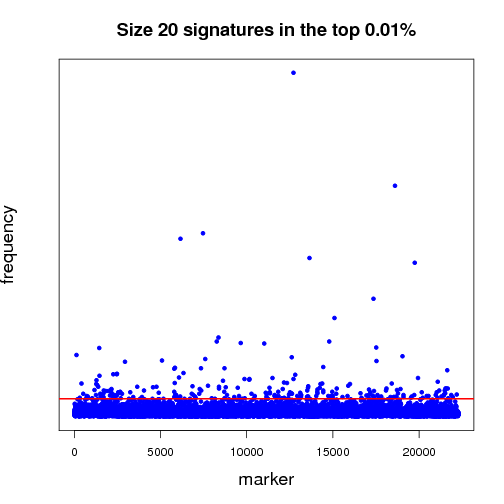
The first half of the “benchmarking” experiment involves determining the performance of markers as the size of the signature changes. For instance, when we compare successful 5-marker signatures against 20-marker signatures, which markers are consistently useful? Which ones increase or diminish in predictive power? Is there an optimum size for signatures? And most importantly, can we identify seemingly minor players that are critical, but not yet in clinical use that can discriminate between normal and disease states?
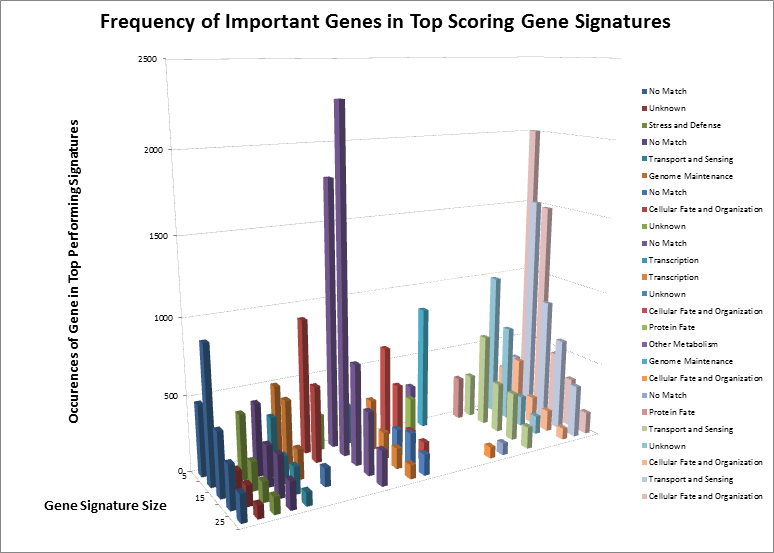
After surveying the first several billion signatures, we have identified the highest-ranking combinations and underlying single genes. After separating those genes by signature size, we can see how some genes remain important regardless of the size, and how other genes “appear” to be important but are only showing up as single events. Considering we have not yet analyzed the complete data set, we have identified the genes by their known functions rather than names, to eliminate any bias towards known markers. However, even by their functions, we can see that many important signaling cascades and biological processes are affected. The most notable of these is “Cellular Fate and Organization”, which makes sense. Sometimes, when an organism loses the ability to naturally kill defective cells, it leads to uncontrolled growth, one of the hallmarks of cancer.
Network Analysis of Major Genes:
To further analyze the nature of our top-performing genes, we can identify their inter-relations in biological networks. We currently maintain one of the largest curated protein-protein interaction databases, which enables us to determine whether our genes (when converted to proteins) are known to interact with other important biomarkers, and in turn, what biological processes may be involved. The graph below shows one such network; nodes in the graph represent genes, edges are physical protein interactions. Node color highlights biological function as described in the legend. Use of biological networks can reveal very small subtleties of how the mechanisms of disease function and elucidate how our proteins may be causing problems; thus, eventually leading to understanding how cancer starts, progresses and how can we treat it.
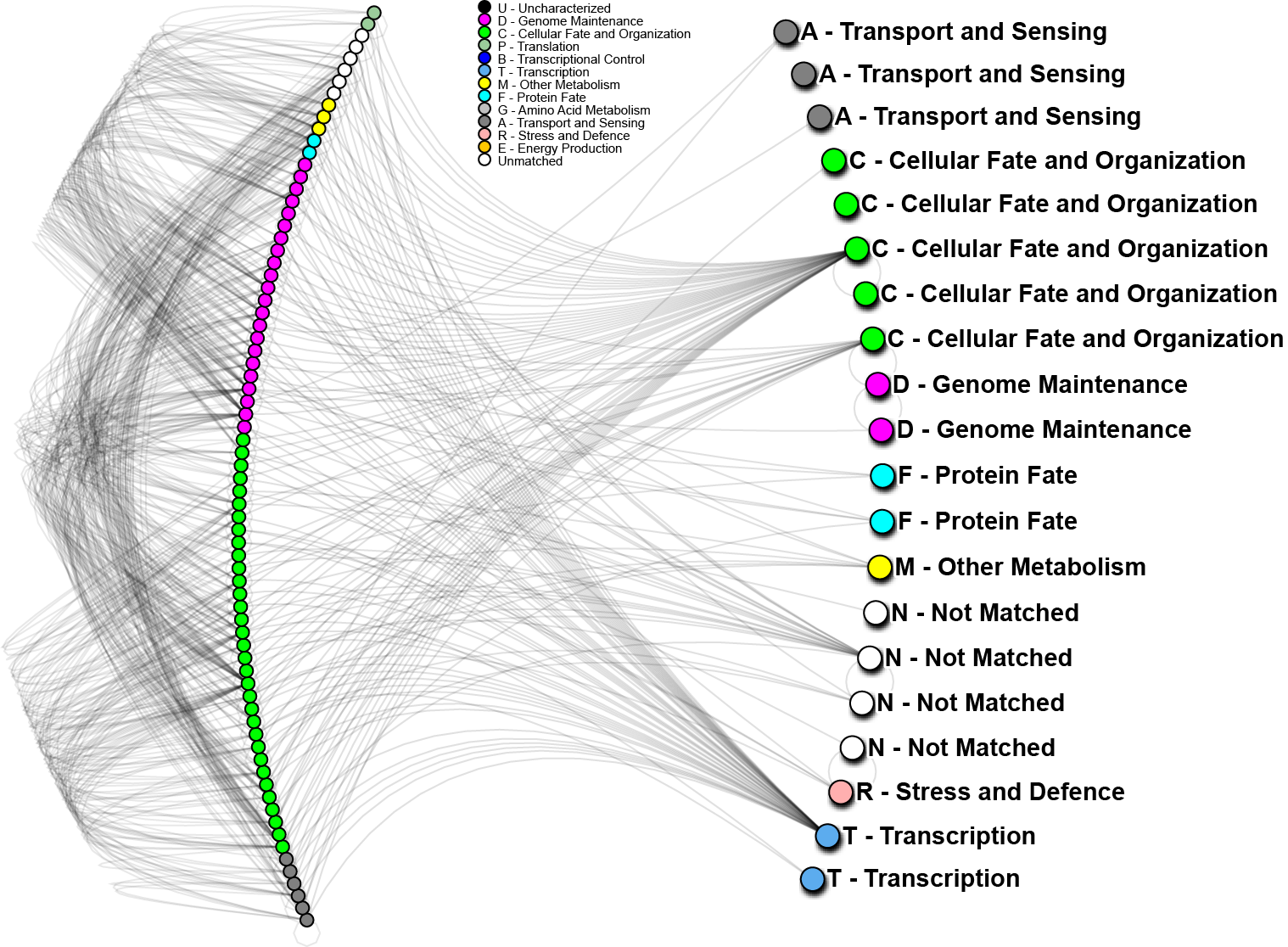
In the above network, 20 out of 24 important proteins we have identified on World Community Grid (right hand side) can be linked through known protein interactions and 56 other proteins (left hand side). We have also conducted a short analysis of the 4 proteins not yet identified using a software prediction package and found those to have significant partners. Those interactions will be evaluated in the near future. The 20 proteins noted above, strikingly, do not interact directly, however, 4 of them show very high interactivity, and can be considered as hubs. From other analyses we know that “hub proteins” are often critical, as they affect many signaling cascades and biological processes. When such proteins malfunction, catastrophic changes often result. On the other hand, proteins with low interactivity could be useful as clinical biomarkers. If they are known to only interact with a few other proteins, then their activity may help to identify particular states of cancer, while having less background “noise”. As a whole we can see that for the most part, our genes of interest are targeting mostly “genome maintenance” and “cellular fate and organization” proteins, which make up about 70% of the interacting proteins (left hand side). This is a good indication that most of the pathways affected are in those major categories, which is consistent with how we understand lung cancer to progress.
Funding & Fundraising:
This past August, we completed our 4th successful Team Ian Ride for Cancer Informatics Research. We were able to raise over $80,000 for cancer research in the name of a former Jurisica student, Ian Van Toch.
Part of this funding is used for the best student paper award at the ISMB conference, and for supporting Cancer Informatics interns.
We also support a special seminar series at Princess Margaret Cancer Center, and the recent presentation by Dr. Wan Lam from BC Cancer Agency discussed “Multi-dimensional Analysis of Lung Cancer Genomes”.